00 - Setup#
import os
import stampede as st
import pandas as pd
import scanpy as sc
import matplotlib.pyplot as plt
from pydeseq2.ds import DeseqStats
from pydeseq2.dds import DeseqDataSet
# dictionary with input files per slide
slides = {
1: {
"exprmat": "exprMat_file.csv.gz",
"metadata": "metadata_file.csv.gz",
"fov_positions": "fov_positions_file.csv.gz",
}
}
# optional filepath prefix
data_dir = "data"
os.makedirs(st.config["adata_dir"], exist_ok=True)
01 - Read#
# table mapping the FOVs per slide to each sample
# column "fovs" accepts any combination of comma separated values (e.g. 1,2,3) and ranges (e.g. 4-6)
samples_df = pd.read_table("data/sample2fov.csv", sep=",")
st.validate_input(slides, samples_df, data_dir)
samples_df
| sample | fovs | slide | |
|---|---|---|---|
| 0 | s1 | 10-18 | 1 |
| 1 | s2 | 19-27 | 1 |
| 2 | s3 | 28-36 | 1 |
# combine the input files into a single cell object and write it to file
adata_file = os.path.join(st.config["adata_dir"], "raw_data.h5ad")
st.read_cosmx(
slides,
samples_df,
adata_file,
data_dir=data_dir,
overwrite=False,
verbose=True,
)
adata_file already exists and overwrite=False
'adatas/raw_data.h5ad'
adata = sc.read_h5ad(adata_file)
adata.obs
| fov | cell_ID | slide | slide-fov | Area | AspectRatio | CenterX_local_px | CenterY_local_px | Width | Height | ... | nCount | nCountPerCell | nFeaturePerCell | propNegativeCellAvg | complexityCellAvg | errorCtPerCellEstimate | percOfDataFromErrorPerCell | qcFlagsFOV | sample | fovs | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| slide-fov-cell_ID | |||||||||||||||||||||
| 1-10-1 | 10 | 1 | 1 | 1-10 | 4260 | 0.97 | 2902 | 613 | 77 | 75 | ... | 140803 | 863.822086 | 670.730061 | 0.000315 | 1.245668 | 87.131902 | 0.100868 | Pass | s1 | 10-18 |
| 1-10-2 | 10 | 2 | 1 | 1-10 | 5228 | 0.95 | 3007 | 623 | 83 | 87 | ... | 140803 | 863.822086 | 670.730061 | 0.000315 | 1.245668 | 87.131902 | 0.100868 | Pass | s1 | 10-18 |
| 1-10-3 | 10 | 3 | 1 | 1-10 | 3204 | 0.89 | 2947 | 649 | 71 | 63 | ... | 140803 | 863.822086 | 670.730061 | 0.000315 | 1.245668 | 87.131902 | 0.100868 | Pass | s1 | 10-18 |
| 1-10-4 | 10 | 4 | 1 | 1-10 | 22940 | 0.81 | 2833 | 746 | 191 | 237 | ... | 140803 | 863.822086 | 670.730061 | 0.000315 | 1.245668 | 87.131902 | 0.100868 | Pass | s1 | 10-18 |
| 1-10-5 | 10 | 5 | 1 | 1-10 | 4404 | 0.76 | 2990 | 691 | 93 | 71 | ... | 140803 | 863.822086 | 670.730061 | 0.000315 | 1.245668 | 87.131902 | 0.100868 | Pass | s1 | 10-18 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 1-36-271 | 36 | 271 | 1 | 1-36 | 4264 | 0.88 | 1812 | 2281 | 81 | 71 | ... | 110510 | 401.854545 | 338.160000 | 0.000438 | 1.168196 | 46.031818 | 0.114548 | Pass | s3 | 28-36 |
| 1-36-272 | 36 | 272 | 1 | 1-36 | 5024 | 0.79 | 397 | 3328 | 87 | 69 | ... | 110510 | 401.854545 | 338.160000 | 0.000438 | 1.168196 | 46.031818 | 0.114548 | Pass | s3 | 28-36 |
| 1-36-273 | 36 | 273 | 1 | 1-36 | 5460 | 0.98 | 323 | 3370 | 83 | 81 | ... | 110510 | 401.854545 | 338.160000 | 0.000438 | 1.168196 | 46.031818 | 0.114548 | Pass | s3 | 28-36 |
| 1-36-274 | 36 | 274 | 1 | 1-36 | 1888 | 0.96 | 455 | 3372 | 47 | 49 | ... | 110510 | 401.854545 | 338.160000 | 0.000438 | 1.168196 | 46.031818 | 0.114548 | Pass | s3 | 28-36 |
| 1-36-275 | 36 | 275 | 1 | 1-36 | 3880 | 0.97 | 398 | 3397 | 69 | 67 | ... | 110510 | 401.854545 | 338.160000 | 0.000438 | 1.168196 | 46.031818 | 0.114548 | Pass | s3 | 28-36 |
27625 rows × 79 columns
02 - QC#
02a - Slide QC#
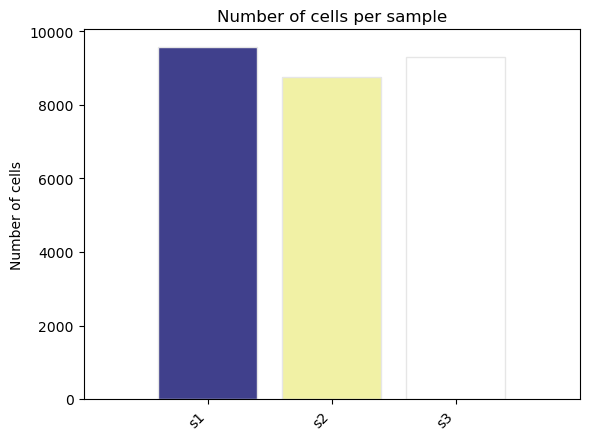
fig, ax = st.pl.ncell_per_condition(adata, "sample")
plt.show()
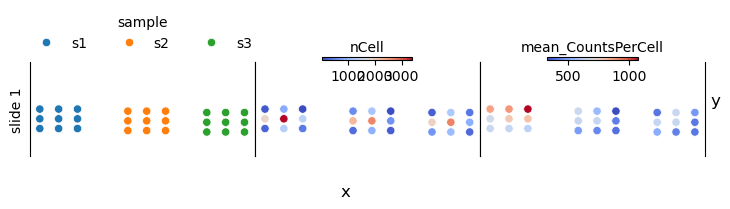
st.pp.slide_qc(adata, slides, ["sample"], data_dir=data_dir)
adata.uns["fov_metadata"].head()
| slide | fov | x | y | nCounts | nCell | mean_CountsPerCell | nCount_negprobes | mean_NegProbe-CountsPerCell | nCount_falsecode | mean_FalseCode-CountsPerCell | mean_CellSize | sample | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| slide-fov | |||||||||||||
| 1-10 | 1 | 10 | 19773 | 149405 | 140803 | 163 | 863.822086 | 46 | 0.282209 | 480 | 2.944785 | 63.957736 | s1 |
| 1-11 | 1 | 11 | 24029 | 149405 | 752088 | 848 | 886.896226 | 274 | 0.323113 | 2569 | 3.029481 | 54.344592 | s1 |
| 1-12 | 1 | 12 | 28285 | 149405 | 50213 | 47 | 1068.361702 | 21 | 0.446809 | 151 | 3.212766 | 74.681880 | s1 |
| 1-13 | 1 | 13 | 19773 | 145149 | 1241593 | 1866 | 665.376742 | 521 | 0.279207 | 5842 | 3.130761 | 52.223075 | s1 |
| 1-14 | 1 | 14 | 24029 | 145149 | 2638265 | 3391 | 778.019758 | 916 | 0.270127 | 10325 | 3.044825 | 52.564838 | s1 |
# this dataset was subset from a much larger slide
# the plot limits in this tutorial are extended to provide a sense of scale
fig, axs = st.pl.slide_qc(
adata, columns=["sample", "nCell", "mean_CountsPerCell"], figsize=(2.5, 0.5), legend_kwargs={"ncols": 3}
)
ylim = axs[1, 0].get_ylim()
axs[1, 0].set_ylim(ylim[0]-10_000, ylim[1]+20_000)
plt.show()
fig, axs = st.pl.slide_qc(
adata, columns=["mean_NegProbe-CountsPerCell", "mean_FalseCode-CountsPerCell", "mean_CellSize"], figsize=(2.5, 0.5)
)
ylim = axs[1, 0].get_ylim()
axs[1, 0].set_ylim(ylim[0]-10_000, ylim[1]+20_000)
plt.show()

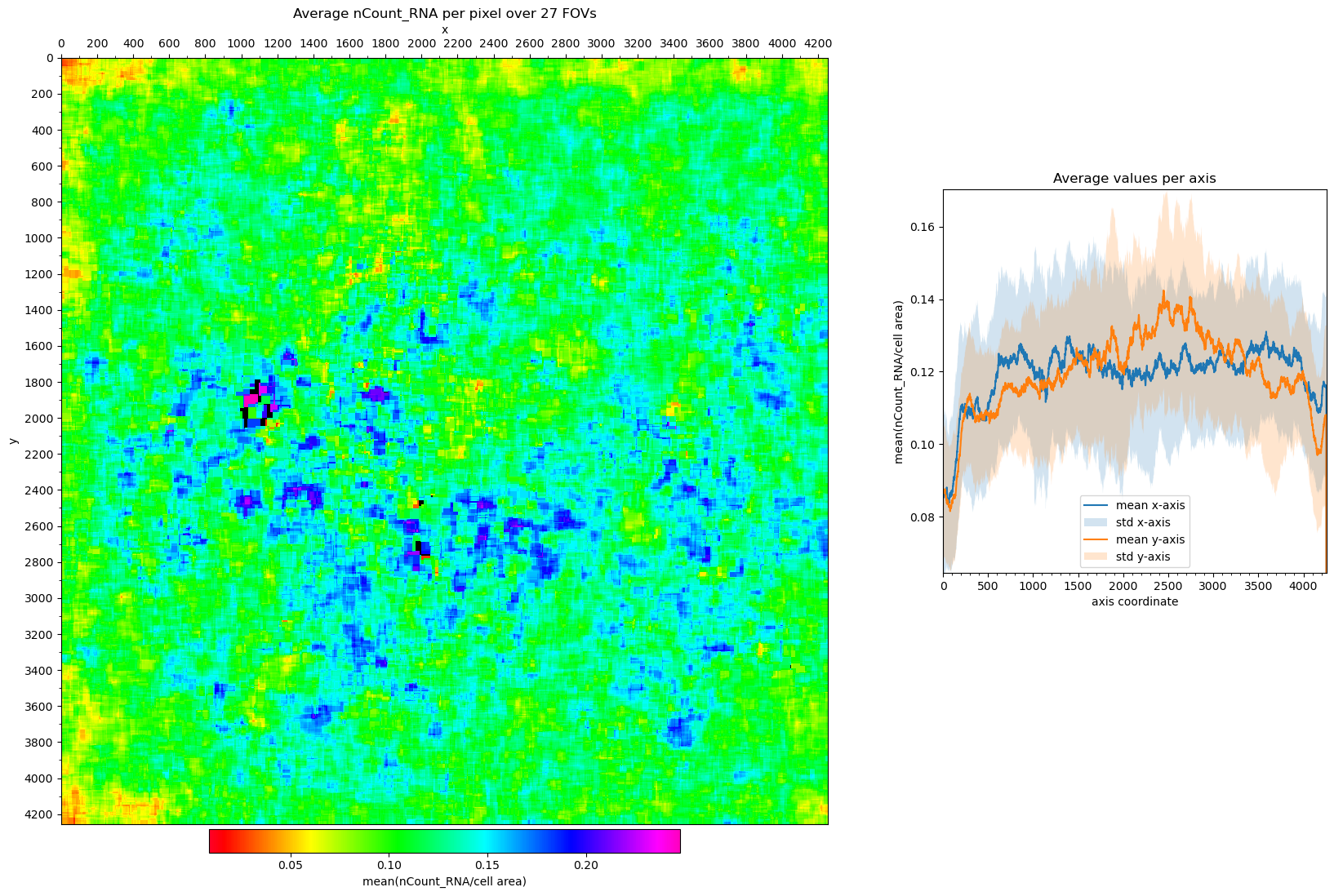
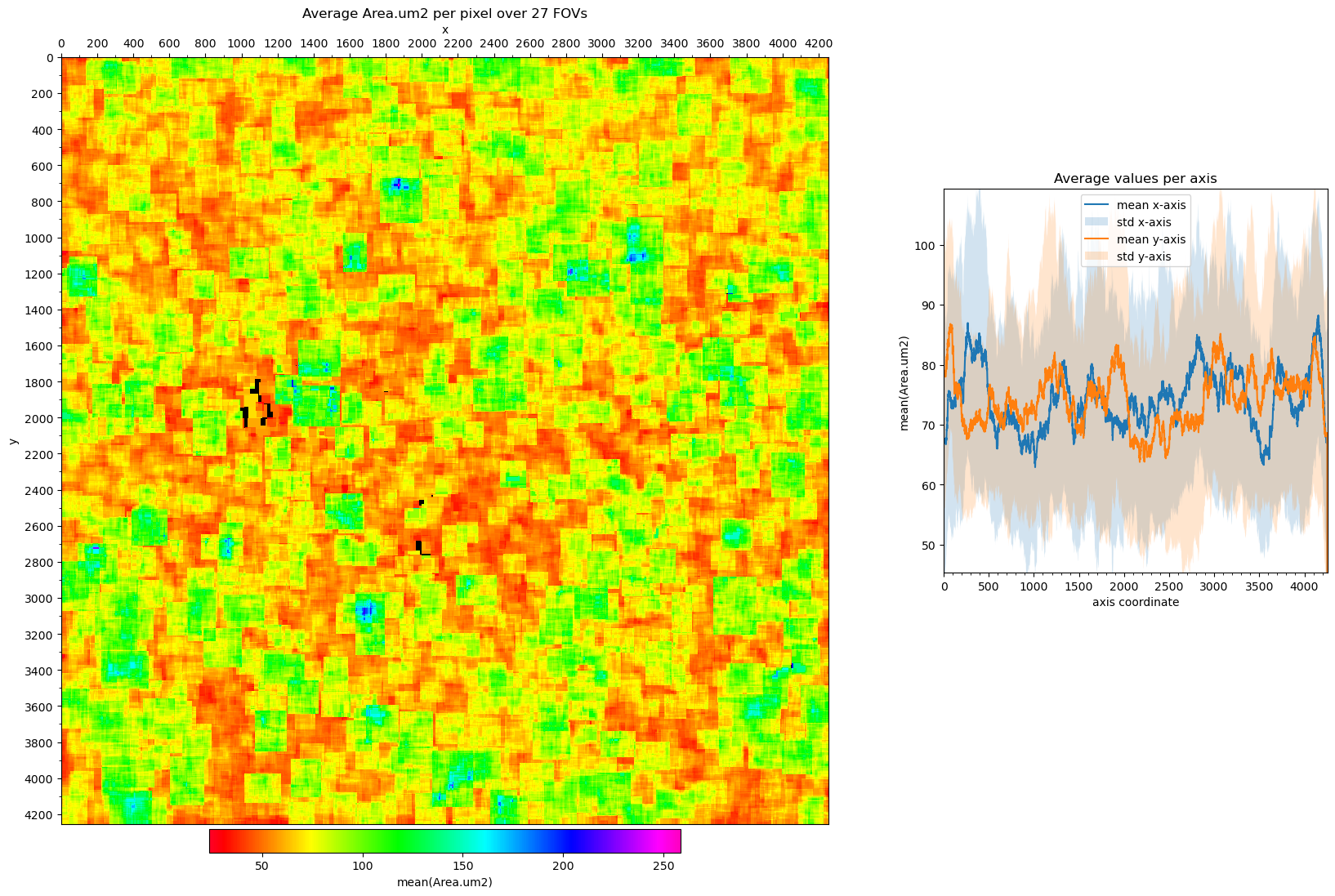
Visualizing Field of View edge-effects#
Cells on the edges of an Field of View (FOV) may have been truncated. Furthermore, we tend to detect less RNA in them. There may also be a bias to the corners and/or one or two edges in the dataset.
# plot the average nCount_RNA per pixel over all FOVs
fig, axs = st.pl.avg_per_pixel(
adata, column='nCount_RNA', normalize_cell_area=True
)
plt.show()
# plot the cell area for all FOVs.
# (do not normalize for cell area when plotting the cell area)
fig, axs = st.pl.avg_per_pixel(
adata, column='Area.um2', normalize_cell_area=False
)
plt.show()
# visualize the effect of removing cells based on distance to FOV edge
fig, axs = st.pl.correlations(adata, xcolumn="dist2edge_px", ycolumn="nCount_RNA", max_quantile=0.7)
dist2edge_threshold = 50
print(f"{dist2edge_threshold=}")
axs[1, 1].axvline(dist2edge_threshold, color="red")
plt.show()
dist2edge_threshold=50
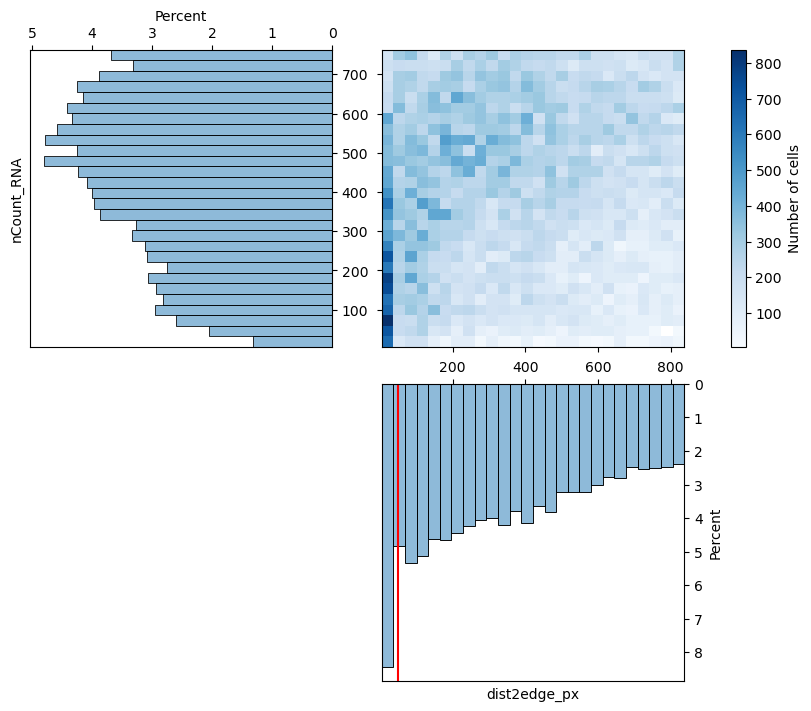
adata = st.pp.filter_edges(adata, dist2edge_threshold)
1_729 cells filtered out, 25_896 cell remaining.
02b - gene QC#
# remove non-PBMC genes
fname = os.path.join(data_dir, "blacklist_pbmc_6k.txt")
blacklist = pd.read_table(fname, header=None)[0].to_list()
before = len(adata.var)
adata = adata[:, ~adata.var.index.isin(blacklist)].copy()
after = len(adata.var)
print(before - after, "genes filtered out")
904 genes filtered out
# mark core immune genes
fname = os.path.join(data_dir, "core_immune_genes_6k.txt")
whitelist = pd.read_table(fname, header=None)[0].to_list()
adata.var["is_core_immune_gene"] = adata.var.index.isin(whitelist)
# determine a noise threshold for gene expression using the mean expression
# and standard deviation of the negative control probes
st.pp.gene_qc(adata)
adata.var
| is_core_immune_gene | is_negctrl | is_sysctrl | nCell | pctCell | nTranscript | mean_Transcript | above_noise | |
|---|---|---|---|---|---|---|---|---|
| A1BG | False | False | False | 5625 | 21.721501 | 6883 | 0.265794 | True |
| A2M | True | False | False | 635 | 2.452116 | 658 | 0.025409 | True |
| AAAS | False | False | False | 1416 | 5.468026 | 1496 | 0.057770 | True |
| AAK1 | True | False | False | 2175 | 8.398981 | 2363 | 0.091250 | True |
| AAMP | False | False | False | 1483 | 5.726753 | 1591 | 0.061438 | True |
| ... | ... | ... | ... | ... | ... | ... | ... | ... |
| ZSWIM6 | True | False | False | 854 | 3.297807 | 888 | 0.034291 | True |
| ZWINT | True | False | False | 969 | 3.741891 | 1019 | 0.039350 | True |
| ZYG11B | False | False | False | 1855 | 7.163268 | 2002 | 0.077309 | True |
| ZYX | True | False | False | 2650 | 10.233241 | 3012 | 0.116311 | True |
| ZZZ3 | True | False | False | 1606 | 6.201730 | 1709 | 0.065995 | True |
5614 rows × 8 columns
fig, ax = st.pl.noise_threshold(adata, max_quantile=0.95)
plt.show()
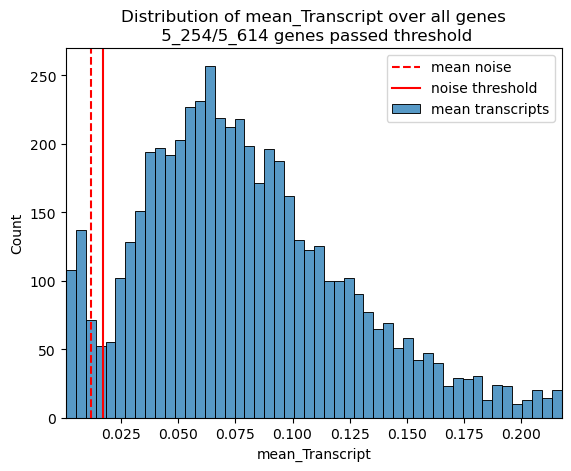
adata = st.pp.filter_genes(adata)
377 genes filtered out, 5_237 genes remaining.
02c - cell QC#
fig, axs = st.pl.violin(
adata,
["nCount_RNA", "nFeature_RNA", "Area.um2", "nCount_negprobes", "nCount_falsecode"],
log_scale=(True, True, True, False, False)
)
plt.show()
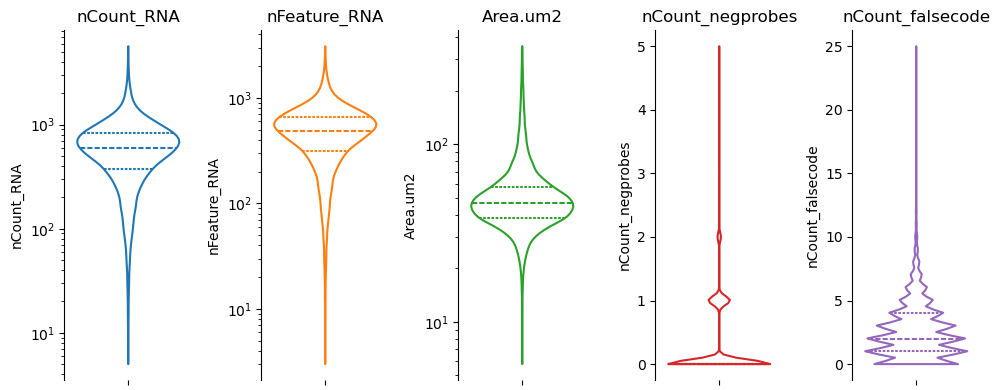
adata = st.pp.filter_cells(
adata,
falsecode_max = 5,
negprobe_max = 2,
ntranscript_min = 250,
ntranscript_max = 1500,
area_min = 25,
area_max = 110,
)
9_123 cells filtered out, 16_773 cells remaining.
st.pp.gene_qc_postfilter(adata)
adata.var[["nCell", "nCell_postfilter", "pctCell", "pctCell_postfilter"]]
| nCell | nCell_postfilter | pctCell | pctCell_postfilter | |
|---|---|---|---|---|
| A1BG | 5625 | 3674 | 21.721501 | 21.90 |
| A2M | 635 | 417 | 2.452116 | 2.49 |
| AAAS | 1416 | 952 | 5.468026 | 5.68 |
| AAK1 | 2175 | 1434 | 8.398981 | 8.55 |
| AAMP | 1483 | 922 | 5.726753 | 5.50 |
| ... | ... | ... | ... | ... |
| ZSWIM6 | 854 | 547 | 3.297807 | 3.26 |
| ZWINT | 969 | 605 | 3.741891 | 3.61 |
| ZYG11B | 1855 | 1180 | 7.163268 | 7.04 |
| ZYX | 2650 | 1766 | 10.233241 | 10.53 |
| ZZZ3 | 1606 | 1067 | 6.201730 | 6.36 |
5237 rows × 4 columns
st.pp.cell_qc_postfilter(adata)
adata.obs[["nFeature_RNA", "nFeature_RNA_postfilter", "nCount_RNA", "nCount_RNA_postfilter"]]
| nFeature_RNA | nFeature_RNA_postfilter | nCount_RNA | nCount_RNA_postfilter | |
|---|---|---|---|---|
| slide-fov-cell_ID | ||||
| 1-10-1 | 627 | 560 | 821 | 748 |
| 1-10-2 | 873 | 771 | 1185 | 1067 |
| 1-10-6 | 648 | 573 | 838 | 748 |
| 1-10-8 | 831 | 716 | 1015 | 888 |
| 1-10-9 | 666 | 583 | 823 | 730 |
| ... | ... | ... | ... | ... |
| 1-36-269 | 750 | 643 | 900 | 786 |
| 1-36-270 | 725 | 629 | 863 | 754 |
| 1-36-272 | 847 | 718 | 1033 | 894 |
| 1-36-273 | 814 | 711 | 989 | 871 |
| 1-36-275 | 660 | 562 | 782 | 676 |
16773 rows × 4 columns
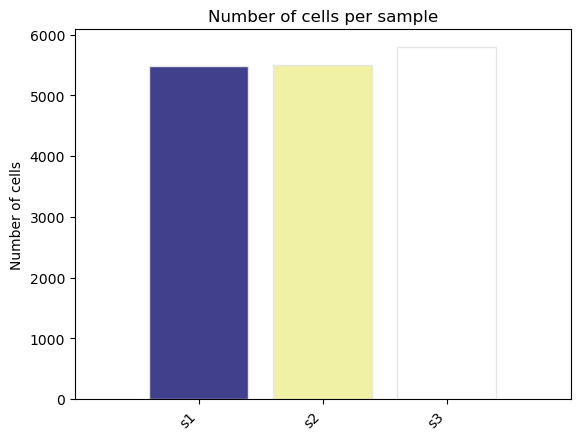
fig, ax = st.pl.ncell_per_condition(adata, columns = ["sample"])
plt.show()
03 - Dimensionality reduction#
st.pp.binarize(adata)
binary layer set as adata.X
st.pp.dim_red(adata, use_genes="is_core_immune_gene")
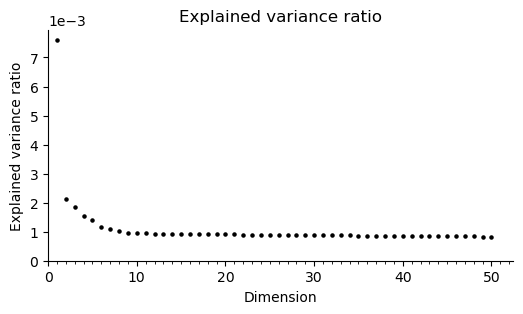
fig, ax = st.pl.scree(adata)
plt.show()
fig_ax_list = st.pl.dim_red(
adata,
columns=["sample"],
subset_size=1000,
)
plt.show()
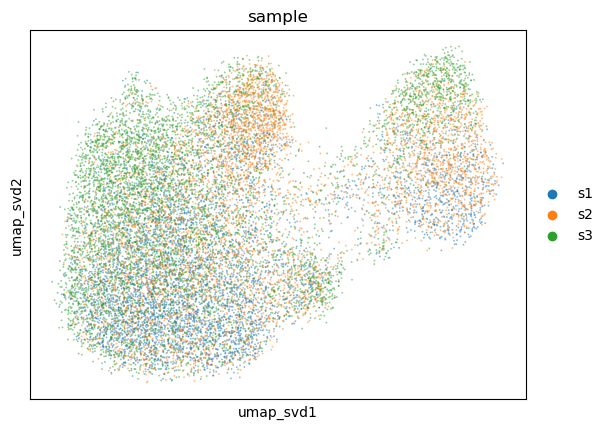
sc.pp.neighbors(
adata,
use_rep="X_svd",
key_added="neighbors_svd",
)
sc.tl.umap(
adata,
neighbors_key="neighbors_svd",
key_added="umap_svd",
)
sc.pl.embedding(
adata,
basis="umap_svd",
color="sample",
alpha=0.5,
)
04 - Clustering#
st.pp.knn_count_smoothing(
adata,
neighbors_key="neighbors_svd",
)
KNN_binary_mean layer set as adata.X
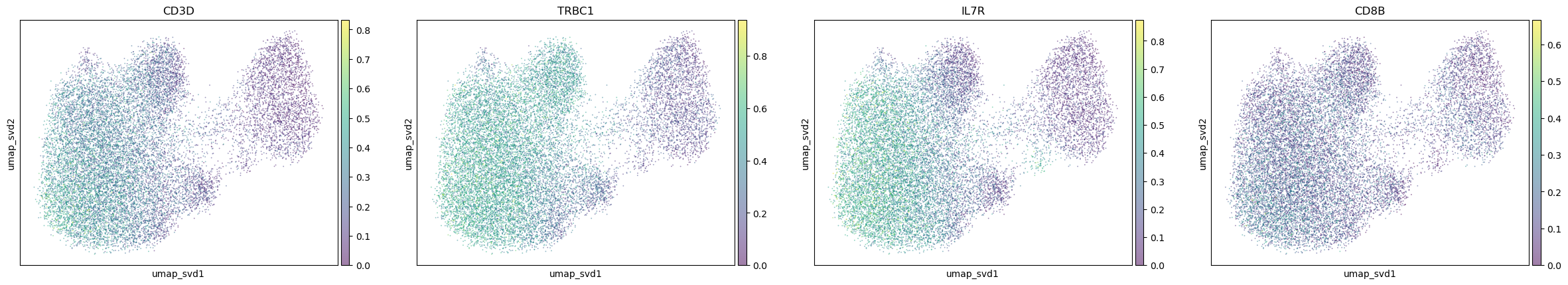
markerdict = {
"T_cells": [
"CD3D", "TRBC1","IL7R", "CD8B",
],
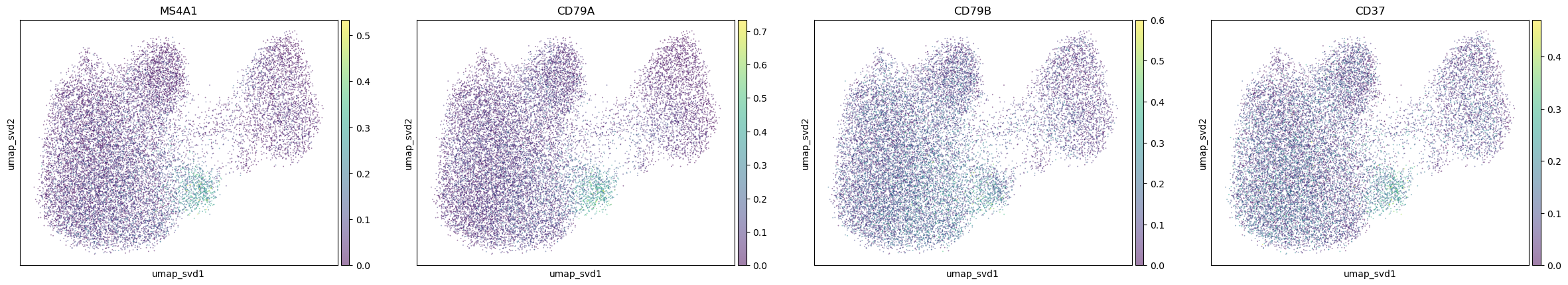
"B_cells": [
"MS4A1", "CD79A", "CD79B", "CD37"
],
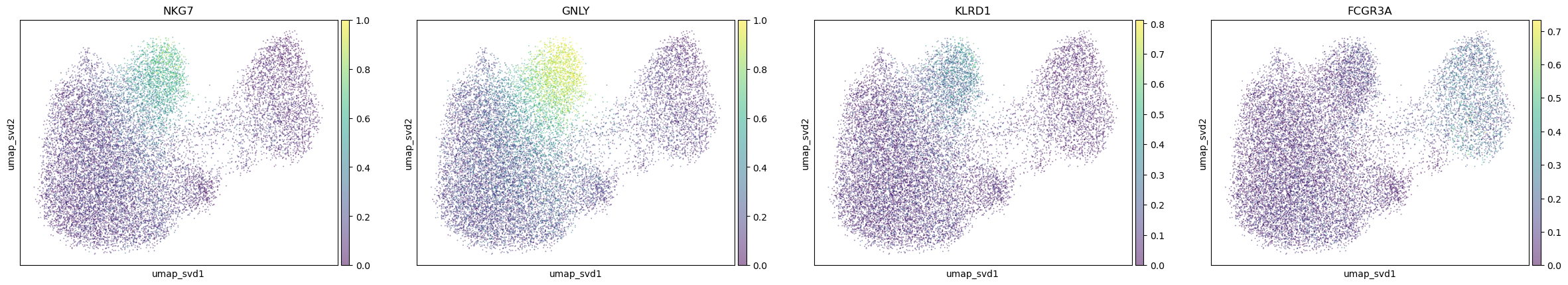
"NK_cells": [
"NKG7", "GNLY", "KLRD1", "FCGR3A"
],
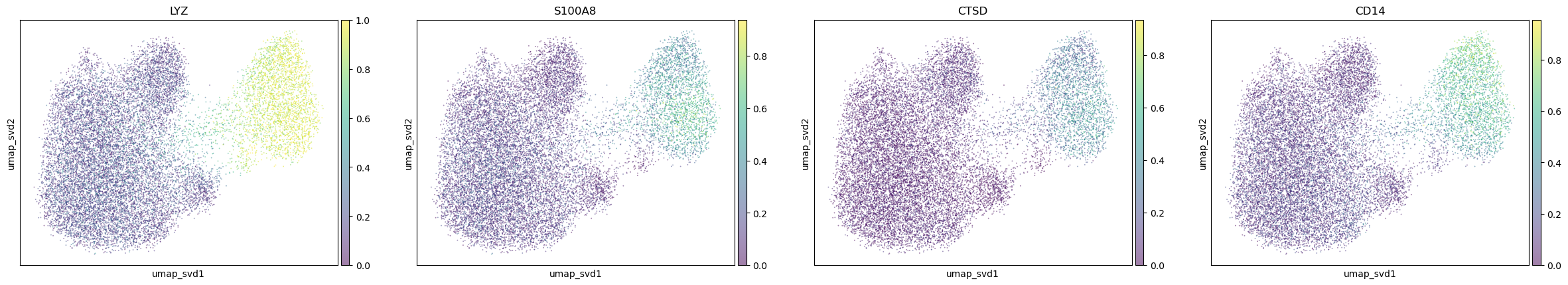
"Monocytes_Macrophages": [
"LYZ", "S100A8", "CTSD", "CD14",
],
}
for ctype, markers in markerdict.items():
print(ctype)
sc.pl.embedding(
adata,
basis="umap_svd",
color=markers,
layer="KNN_binary_mean",
ncols=4,
alpha=0.5,
)
T_cells
B_cells
NK_cells
Monocytes_Macrophages
res = 0.5
sc.tl.leiden(
adata,
resolution=res,
neighbors_key="neighbors_svd",
key_added=f"leiden_res_{res:.2f}",
flavor="igraph",
n_iterations=2,
directed=False,
random_state=42,
)
sc.tl.rank_genes_groups(adata, f"leiden_res_{res:.2f}", n_genes=6, layer="binary")
sc.tl.dendrogram(adata, f"leiden_res_{res:.2f}", use_rep="X_svd")
sc.pl.rank_genes_groups_matrixplot(
adata,
n_genes=6,
)
WARNING: It seems you use rank_genes_groups on the raw count data. Please logarithmize your data before calling rank_genes_groups.
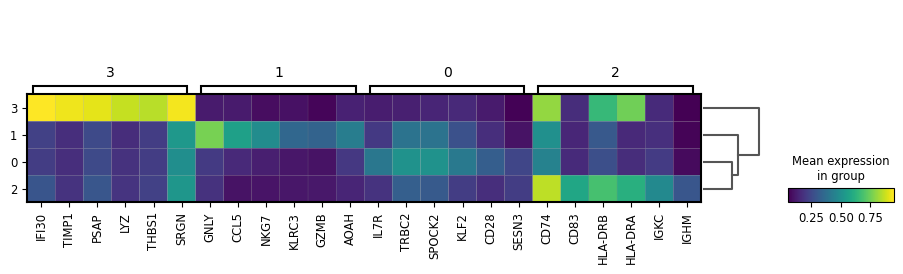
cluster2ctype = {
"0": "T_cells",
"1": "NK_cells",
"2": "B_cells",
"3": "Myeloid_cells",
}
adata.obs["celltype_lvl1"] = adata.obs[f"leiden_res_{res:.2f}"].astype(str).replace(cluster2ctype).astype("category")
sc.pl.embedding(
adata,
basis="umap_svd",
color="celltype_lvl1",
alpha=0.5,
)
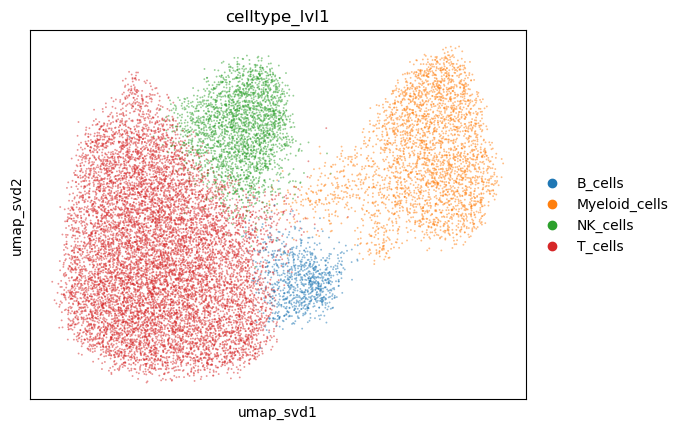
05 - Differential expression analysis#
st.pp.combine_obs_columns(adata, ["sample", "celltype_lvl1"], "sample_celltype")
adata.obs["sample_celltype"].value_counts().head()
sample_celltype
s1_T_cells 4003
s3_T_cells 3912
s2_T_cells 2483
s2_NK_cells 1359
s2_Myeloid_cells 1351
Name: count, dtype: int64
05a - using detection rates & pbGLM#
det_rate_df = st.pp.detection_rates(adata, "sample_celltype")
det_rate_df
| s1_T_cells | s3_T_cells | s2_T_cells | s2_NK_cells | s2_Myeloid_cells | s3_Myeloid_cells | s1_Myeloid_cells | s3_NK_cells | s1_NK_cells | s3_B_cells | s2_B_cells | s1_B_cells | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A1BG | 0.166938 | 0.186624 | 0.192226 | 0.180092 | 0.241642 | 0.234460 | 0.198680 | 0.152472 | 0.154464 | 0.222320 | 0.177528 | 0.168530 |
| A2M | 0.022996 | 0.016689 | 0.019951 | 0.024067 | 0.019234 | 0.015267 | 0.023463 | 0.016206 | 0.023768 | 0.027226 | 0.022711 | 0.013955 |
| AAAS | 0.046860 | 0.046575 | 0.053780 | 0.039238 | 0.046576 | 0.037077 | 0.039669 | 0.037278 | 0.053029 | 0.051782 | 0.071536 | 0.051186 |
| AAK1 | 0.076153 | 0.075050 | 0.077622 | 0.067599 | 0.053272 | 0.056707 | 0.068781 | 0.092419 | 0.077461 | 0.051782 | 0.042722 | 0.019593 |
| AAMP | 0.044045 | 0.040120 | 0.048625 | 0.032338 | 0.047914 | 0.047983 | 0.065840 | 0.038899 | 0.047833 | 0.049051 | 0.071536 | 0.059978 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| ZSWIM6 | 0.026101 | 0.020126 | 0.022668 | 0.024756 | 0.045907 | 0.041439 | 0.040629 | 0.021068 | 0.022061 | 0.027226 | 0.017018 | 0.030966 |
| ZWINT | 0.032164 | 0.033675 | 0.026749 | 0.031648 | 0.018569 | 0.032715 | 0.025359 | 0.019447 | 0.032328 | 0.032678 | 0.028416 | 0.019593 |
| ZYG11B | 0.061227 | 0.052116 | 0.061703 | 0.058594 | 0.064679 | 0.061069 | 0.054143 | 0.056735 | 0.056500 | 0.057245 | 0.071536 | 0.028111 |
| ZYX | 0.064447 | 0.086215 | 0.068612 | 0.069678 | 0.139921 | 0.201745 | 0.144557 | 0.113513 | 0.068700 | 0.076388 | 0.048464 | 0.068847 |
| ZZZ3 | 0.050390 | 0.053733 | 0.058255 | 0.052366 | 0.044569 | 0.050164 | 0.055113 | 0.058357 | 0.046104 | 0.068180 | 0.062864 | 0.036702 |
5237 rows × 12 columns
%%time
results_df1 = st.tl.paired_binomial_glm(
det_rate_df,
adata,
column="sample_celltype",
test_condition="NK_cells",
reference_condition="Myeloid_cells",
condition_column="celltype_lvl1",
)
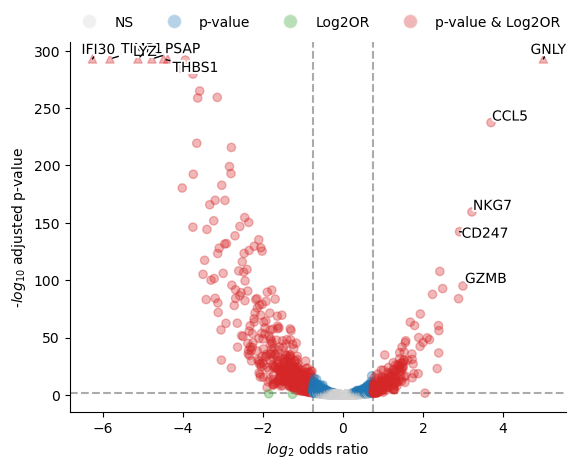
results_df1
CPU times: user 22.3 s, sys: 15.9 ms, total: 22.3 s
Wall time: 22.3 s
| beta | se | odds_ratio | pval | perfect_separation | error | padj | -log10(padj) | log2(odds_ratio) | |
|---|---|---|---|---|---|---|---|---|---|
| gene | |||||||||
| IFI30 | -4.339651 | 0.090796 | 0.013041 | 0.000000e+00 | False | None | 0.000000e+00 | 9.0 | -6.260793 |
| TIMP1 | -4.036057 | 0.084280 | 0.017667 | 0.000000e+00 | False | None | 0.000000e+00 | 9.0 | -5.822799 |
| LYZ | -3.551084 | 0.079467 | 0.028694 | 0.000000e+00 | False | None | 0.000000e+00 | 9.0 | -5.123131 |
| PSAP | -3.309378 | 0.073933 | 0.036539 | 0.000000e+00 | False | None | 0.000000e+00 | 9.0 | -4.774423 |
| THBS1 | -3.108490 | 0.072474 | 0.044668 | 0.000000e+00 | False | None | 0.000000e+00 | 9.0 | -4.484603 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| CD247 | 2.019324 | 0.078699 | 7.533233 | 3.378460e-145 | False | None | 5.529061e-143 | 9.0 | 2.913269 |
| GZMB | 2.077770 | 0.099078 | 7.986641 | 1.206773e-97 | False | None | 1.128548e-95 | 9.0 | 2.997589 |
| NKG7 | 2.233148 | 0.082191 | 9.329190 | 1.460050e-162 | False | None | 3.058512e-160 | 9.0 | 3.221752 |
| CCL5 | 2.563929 | 0.077387 | 12.986745 | 1.047344e-240 | False | None | 3.917816e-238 | 9.0 | 3.698968 |
| GNLY | 3.470331 | 0.078625 | 32.147383 | 0.000000e+00 | False | None | 0.000000e+00 | 9.0 | 5.006629 |
5237 rows × 9 columns
fig, ax = st.pl.paired_binomial_glm_volcano(results_df1)
plt.show()
05b - using pyDESeq2#
counts = st.pp.pseudobulk(adata, "sample_celltype")
counts
| s1_T_cells | s1_Myeloid_cells | s1_NK_cells | s1_B_cells | s2_T_cells | s2_Myeloid_cells | s2_NK_cells | s2_B_cells | s3_T_cells | s3_B_cells | s3_Myeloid_cells | s3_NK_cells | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A1BG | 917 | 198 | 88 | 56 | 550 | 355 | 259 | 61 | 800 | 81 | 215 | 94 |
| A2M | 135 | 25 | 14 | 5 | 59 | 29 | 35 | 8 | 73 | 10 | 14 | 10 |
| AAAS | 272 | 42 | 31 | 18 | 158 | 70 | 57 | 25 | 203 | 19 | 34 | 23 |
| AAK1 | 436 | 72 | 45 | 7 | 227 | 80 | 98 | 15 | 326 | 19 | 52 | 57 |
| AAMP | 256 | 69 | 28 | 21 | 143 | 72 | 47 | 25 | 175 | 18 | 44 | 24 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| ZSWIM6 | 153 | 43 | 13 | 11 | 67 | 69 | 36 | 6 | 88 | 10 | 38 | 13 |
| ZWINT | 188 | 27 | 19 | 7 | 79 | 28 | 46 | 10 | 147 | 12 | 30 | 12 |
| ZYG11B | 353 | 57 | 33 | 10 | 181 | 97 | 85 | 25 | 227 | 21 | 56 | 35 |
| ZYX | 371 | 147 | 40 | 24 | 201 | 208 | 101 | 17 | 374 | 28 | 185 | 70 |
| ZZZ3 | 292 | 58 | 27 | 13 | 171 | 67 | 76 | 22 | 234 | 25 | 46 | 36 |
5237 rows × 12 columns
%%time
results_df2 = st.tl.pydeseq2(
counts,
adata,
column="sample_celltype",
test_condition="NK_cells",
reference_condition="Myeloid_cells",
condition_column="celltype_lvl1",
)
results_df2
Using None as control genes, passed at DeseqDataSet initialization
Fitting size factors...
... done in 0.00 seconds.
Fitting dispersions...
... done in 0.51 seconds.
Fitting dispersion trend curve...
/home/siebrenf/miniconda3/envs/stampede/lib/python3.14/site-packages/pydeseq2/dds.py:822: UserWarning: The dispersion trend curve fitting did not converge. Switching to a mean-based dispersion trend.
self._fit_parametric_dispersion_trend(vst)
... done in 0.11 seconds.
Fitting MAP dispersions...
... done in 0.51 seconds.
Fitting LFCs...
... done in 0.51 seconds.
Calculating cook's distance...
... done in 0.01 seconds.
Replacing 0 outlier genes.
Running Wald tests...
Log2 fold change & Wald test p-value: celltype_lvl1 NK_cells vs Myeloid_cells
baseMean log2FoldChange lfcSE stat pvalue padj
A1BG 200.158056 -0.439831 0.150883 -2.915053 0.003556 0.021761
A2M 22.222461 0.166992 0.297768 0.560814 0.574924 0.787363
AAAS 51.730993 0.011885 0.219241 0.054211 0.956767 0.982888
AAK1 68.129857 0.377736 0.189506 1.993267 0.046232 0.170509
AAMP 53.521286 -0.496203 0.227030 -2.185632 0.028843 0.120262
... ... ... ... ... ... ...
ZSWIM6 30.811360 -0.894070 0.255361 -3.501200 0.000463 0.003743
ZWINT 30.131072 0.201892 0.270985 0.745032 0.456252 0.698725
ZYG11B 61.445199 -0.070701 0.197421 -0.358122 0.720252 0.875431
ZYX 103.154574 -0.952321 0.226699 -4.200818 0.000027 0.000275
ZZZ3 57.277142 0.076049 0.202864 0.374878 0.707751 0.869661
[5237 rows x 6 columns]
CPU times: user 3.09 s, sys: 120 ms, total: 3.21 s
Wall time: 4.09 s
... done in 0.46 seconds.
| baseMean | log2FoldChange | lfcSE | stat | pvalue | padj | |
|---|---|---|---|---|---|---|
| A1BG | 200.158056 | -0.439831 | 0.150883 | -2.915053 | 0.003556 | 0.021761 |
| A2M | 22.222461 | 0.166992 | 0.297768 | 0.560814 | 0.574924 | 0.787363 |
| AAAS | 51.730993 | 0.011885 | 0.219241 | 0.054211 | 0.956767 | 0.982888 |
| AAK1 | 68.129857 | 0.377736 | 0.189506 | 1.993267 | 0.046232 | 0.170509 |
| AAMP | 53.521286 | -0.496203 | 0.227030 | -2.185632 | 0.028843 | 0.120262 |
| ... | ... | ... | ... | ... | ... | ... |
| ZSWIM6 | 30.811360 | -0.894070 | 0.255361 | -3.501200 | 0.000463 | 0.003743 |
| ZWINT | 30.131072 | 0.201892 | 0.270985 | 0.745032 | 0.456252 | 0.698725 |
| ZYG11B | 61.445199 | -0.070701 | 0.197421 | -0.358122 | 0.720252 | 0.875431 |
| ZYX | 103.154574 | -0.952321 | 0.226699 | -4.200818 | 0.000027 | 0.000275 |
| ZZZ3 | 57.277142 | 0.076049 | 0.202864 | 0.374878 | 0.707751 | 0.869661 |
5237 rows × 6 columns
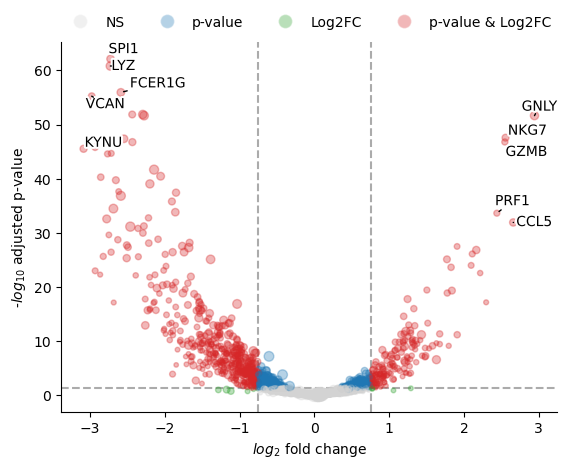
fig, axs = st.pl.pydeseq2_volcano(results_df2)
plt.show()